研究内容Research
We study local structures of non-crystalline metal alloys (LPSO-Mg alloys, Metallic glasses), interactions between atoms/clusters in alloys, electronic/magnetic properties and atomic structures of nano-materials including transition metal elements , such as metal nano-clusters and metal clusters on nano-tubes by using first principles calculation based on density functional theory.
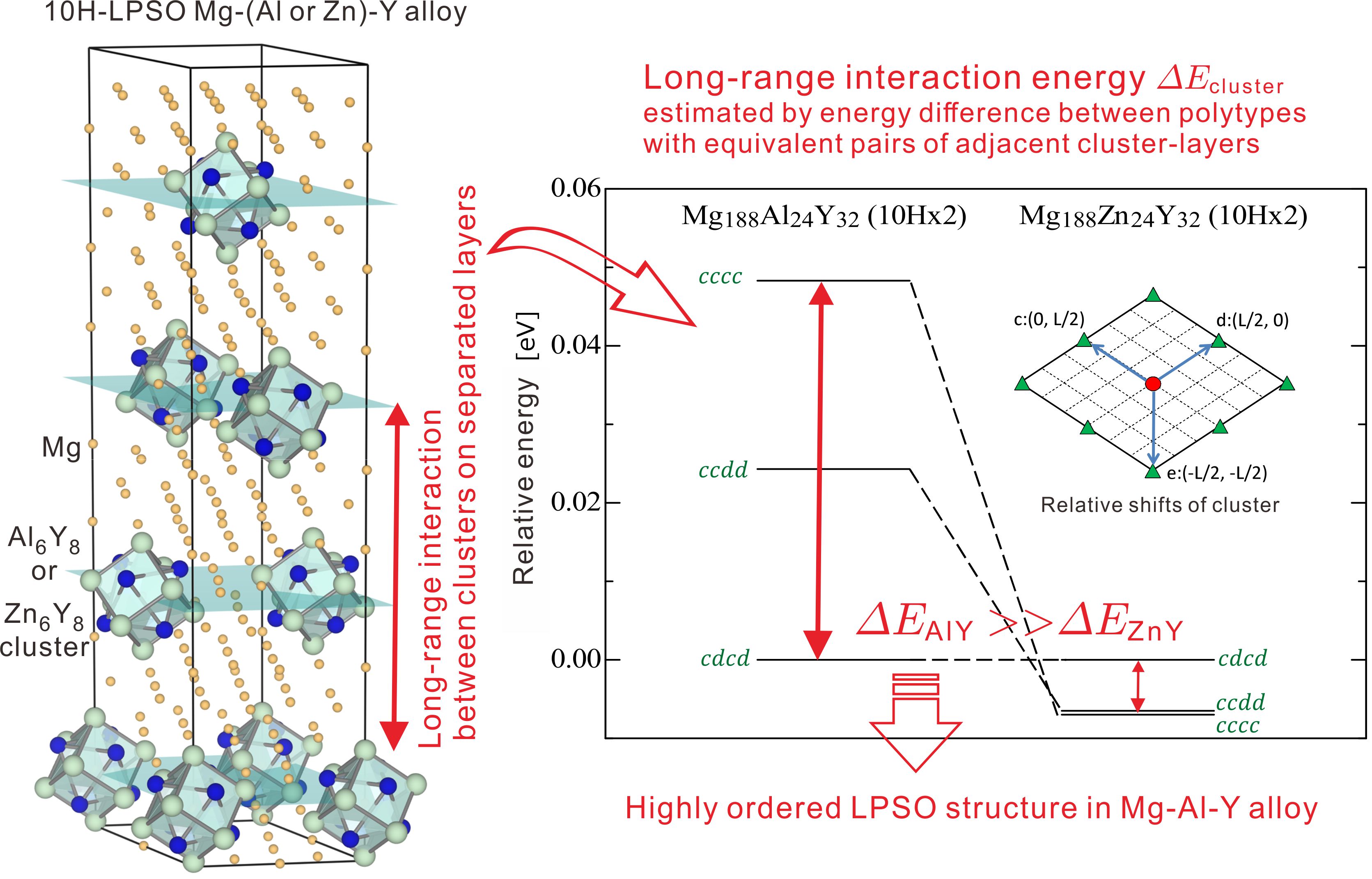
Long-Period Stacking Ordered(LPSO: Millefeuille) structure and Interactons between solute clusters in Mg alloys
Long-Period Stacking Ordered(LPSO) Mg alloys have attracted much attention as new light-weight, high-strength and high heat-resistance structural materials for transport machinery. The LPSO-Mg alloys have an anomalous structure, where solute elements such as Al, Zn, Y are incrassated in the periodic stacking fault layers and constitute L12-type clusters. These structures of the stacking fault layers (Millefeuille structures) and interaction between the solute clusters are keys to high-strength Mg alloys. We calculate long-range interactions between Al-Y (Zn-Y) clusters along the stacking direction in Mg-Al(Zn)-Y alloys with long-period stacking ordered (LPSO) structure by using first-principles calculation based on density functional theory. We reveal that the long-range interactions in the Mg-Al-Y alloys are one digit larger than thosein the Mg-Zn-Y alloys. These results suggest that the long-range interactions between Al-Y clusters play an important role to form the highly ordered LPSO structure in Mg-Al-Rare earth alloys.
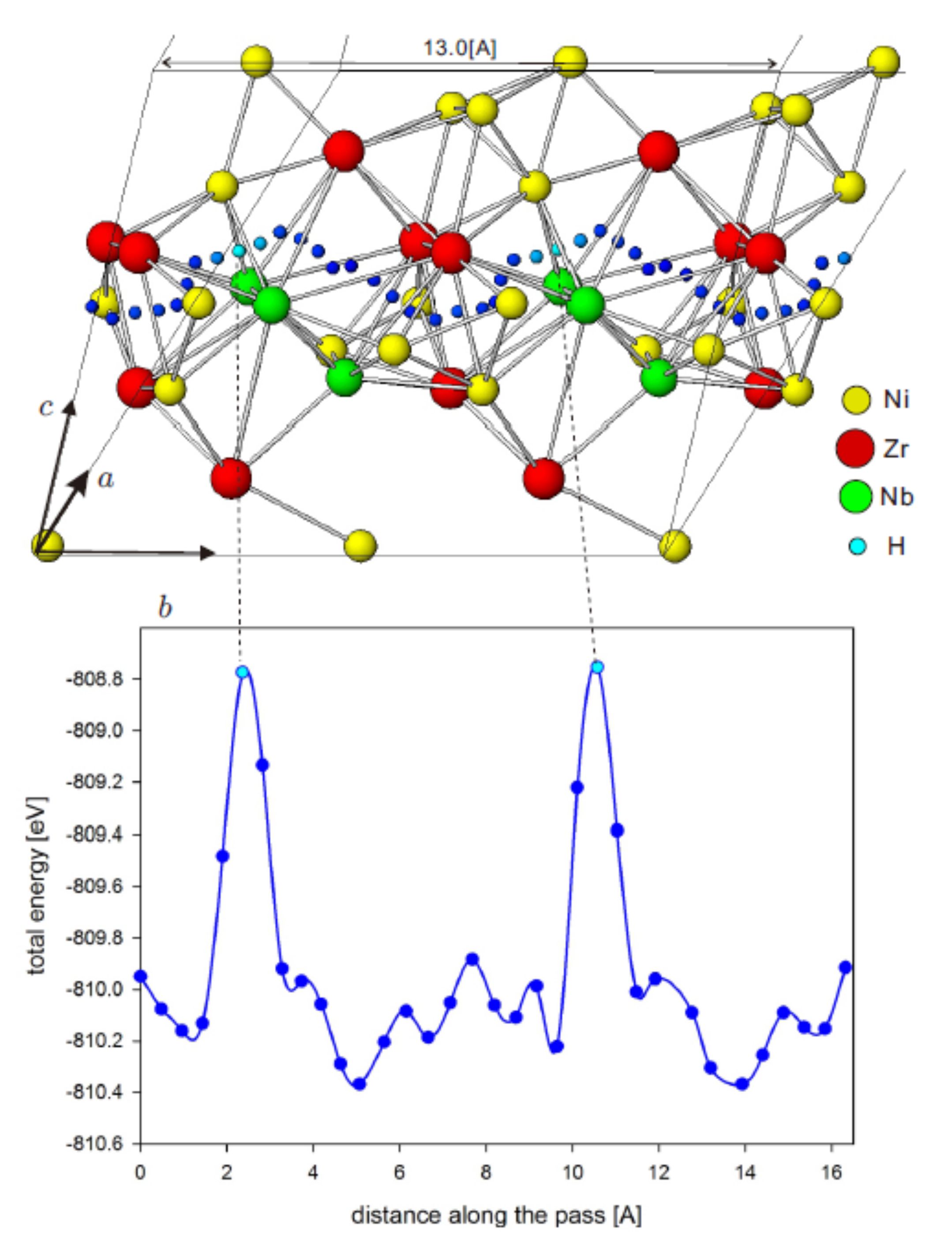
Proton tranport in Zr-Ni-Nb amorphous alloy
Transition-metal amorphous alloys are expected to be applied to hydrogen energy systems. Energetically favorable sites and diffusion paths for hydrogen atom in the local structure of amorphous alloys and the structural change by introducing hydrogen atoms give fundamental information to understand hydrogen storage property, permeability and embrittlement of the amorphous alloys as materials for hydrogen energy system.
We study optimized atomic sites, minimum energy paths for hydrogen atom in Zr-Ni-Nb amorphous alloys, and optimized local structure of the hydrogenized alloy by using the first principles calculation to clarify (1) thermal and kinetic properties for hydrogen atom, such as activation energy on the diffusion path (left-hand side figure), (2) feasible diffusion process of hydrogen atom ,and (3) change of permeability and embrittlement process in the Zr-Ni-Nb amorphous alloy.
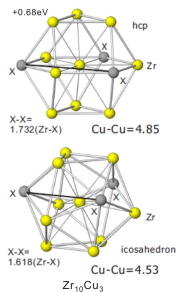
Stablity of local structure in Metal glasses
Metal glasses (MG) known as amorphous materials both with high hardness and with high toughness and are applied to various devises. These anomalous properties are originatedfrom local nano-structural units constructing MG. It is important for new material design of MG to elucidate the mechanism of stability of the local nano-structure in MD.
We study the stability of icosahedral cluster, Zr10Cu3, in the Zr-Cu MD by calculating many-body interaction energy between Zr and Cu with on the full potential KKR method. The Left-hand side figure shows that the icosahedral Zr10Cu cluster is more stable than the hcp cluster. The stability is explained by comparing 2-body interaction energies for the icosahedral cluster with those for the hcp cluster.
We also construct model potential for MG, which reproduces the many-body interaction energy by the ab initio calculation, to be used in the large size molecular dynamics.
We study the stability of icosahedral cluster, Zr10Cu3, in the Zr-Cu MD by calculating many-body interaction energy between Zr and Cu with on the full potential KKR method. The Left-hand side figure shows that the icosahedral Zr10Cu cluster is more stable than the hcp cluster. The stability is explained by comparing 2-body interaction energies for the icosahedral cluster with those for the hcp cluster.
We also construct model potential for MG, which reproduces the many-body interaction energy by the ab initio calculation, to be used in the large size molecular dynamics.
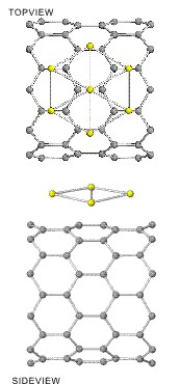
Atomic structure and property of transition-metal cluster on carbon nano-tube
Carbon nano-tubes (CNT) covered with transition-metal (TM) have high reactivity to various gases and are expected to be applied to CO-sensor etc. The structure of TM materials are depends on the element: Ti atoms 2-dimensionaly cover the surface while Gold atoms aggregate into a 3-dimensional cluster.
To understand these properties, gas-reactivity, structure of TM material, it is needed to study not only the single atom properties on the various atomic sites on CNT but also the properties as TM cluster, i.e. influence of binding energy between TM atoms, bond-length mismatch between C-C and TM-TM bonds, etc., upon the stability and gas-reactivity of TM cluster.
We study the optimized structure and electronic states of TM clusters on the various sites of CNT by using the first principles calculation to elucidate the size/element dependence of electronic and geometrical structures. The left-hand side figure shows the optimized structure of hexagonal Ti7 cluster on zigzag type (10,0) CNT.
To understand these properties, gas-reactivity, structure of TM material, it is needed to study not only the single atom properties on the various atomic sites on CNT but also the properties as TM cluster, i.e. influence of binding energy between TM atoms, bond-length mismatch between C-C and TM-TM bonds, etc., upon the stability and gas-reactivity of TM cluster.
We study the optimized structure and electronic states of TM clusters on the various sites of CNT by using the first principles calculation to elucidate the size/element dependence of electronic and geometrical structures. The left-hand side figure shows the optimized structure of hexagonal Ti7 cluster on zigzag type (10,0) CNT.
Fujima Lab.藤間研究室
Fac. Engineering Bld.6 3F307
Shizuoka Univ. 3-5-1 Naka-ku Johoku Hamamatsu 432-8561